Researches
Deep Learning Quantum Monte Carlo
The deep learning quantum Monte Carlo (QMC) method provides new opportunities for solving electronic structure of molecules and materials, where the wavefunction is described by a neural network optimized based on the variational principle. This method has shown great promise in accurately determining ground-state and excited state wavefunctions, even for strongly-correlated systems, while keeping computational costs manageable.
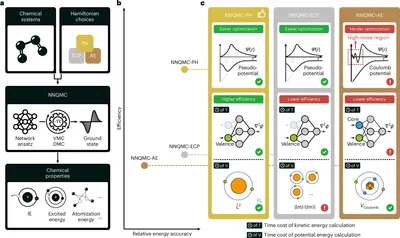
Pseudo-Hamiltonian for NNQMC study of large quantum many-body systems
Computing the quantum behavior of interacting electrons is central to chemistry and physics, but scaling wavefunction simulations to large, strongly correlated systems remains computationally prohibitive. While neural network-based quantum Monte Carlo (NNQMC) shows great promise, conventional semi-local pseudopotentials introduce expensive nonlocal terms that severely bottleneck efficiency. We integrate fully local pseudopotentials (pseudo-Hamiltonians) into NNQMC and apply them to main-group elements and complex iron–sulfur clusters. By eliminating costly nonlocal integrations, our approach accelerates NNQMC by over an order of magnitude while preserving high accuracy, unlocking the simulation of previously inaccessible strongly correlated systems.
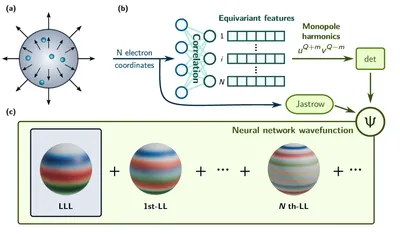
DeepHall framework for QMC study of fractional quantum Hall system
Strong correlation drives exotic emergent phenomena, challenging to model—fractional quantum Hall systems being a key example. Traditional methods often neglect Landau level mixing due to its complexity, even though this mixing can significantly alter the ground state. We propose the DeepHall deep learning framework and apply it to FQH systems at 1/3 and 2/5 fillings, naturally including higher Landau levels and achieving lower energies than lower Landau level exact diagonalization, matching state-of-the-art methods like fp-DMC. This highlights deep learning’s potential to advance studies of strongly correlated systems.
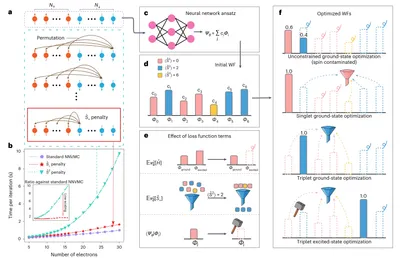
Spin-symmetry-enforced deep learning QMC
Optimizations in deep learning QMC methods are not guaranteed to produce quantum states with the desired spin symmetry. We introduce a low-scaling penalty term that enforces spin symmetry with minimal computational overhead, achieving higher accuracy and efficiency for excited-state calculations.
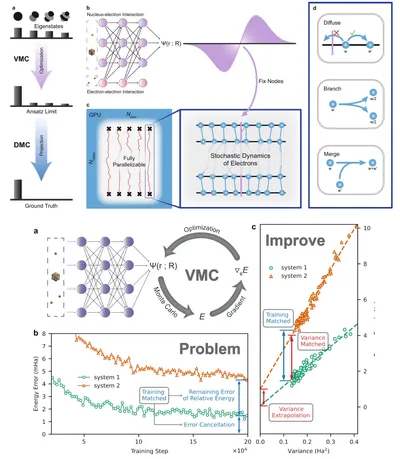
Deep learning diffusion Monte Carlo and variance extrapolation
The accuracy of deep learning QMC energy results depends on the neural network’s parameters and ansatz form. To further improve this, we combine deep learning QMC with diffusion Monte Carlo (DMC), leveraging the accurate wavefunction node structures from deep learning to achieve lower variational energy. Additionally, we identified a linear relationship between energy and local energy variance during training, enabling energy extrapolation. This significantly improves accuracy, with extrapolated relative energies aligning better with experimental results.
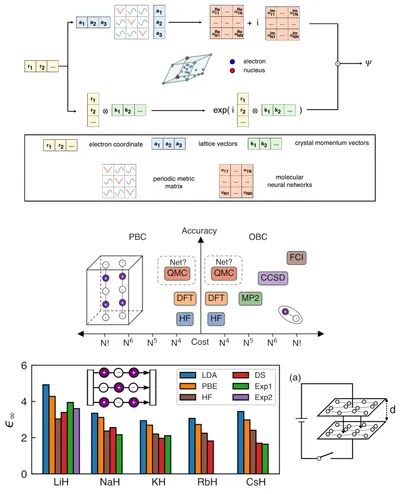
DeepSolid framework for QMC study of solids
Our DeepSolid framework enables deep learning QMC modeling of periodic systems, achieving excellent energy consistency with state-of-the-art methods. Beyond energy, DeepSolid addresses the challenge of calculating electric susceptibility in solids, a task hindered by the theoretical complexity of the Berry phase. While DFT remains the most common method, DeepSolid provides a robust and high-accuracy approach for electronic structures.
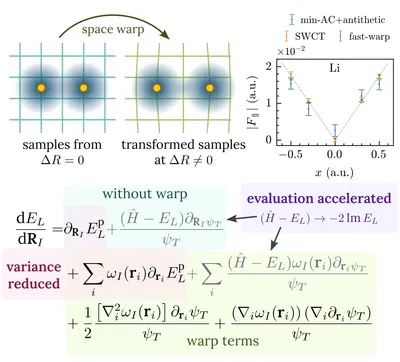
Interatomic force calculation with deep learning QMC
While most developments in deep learning QMC focus on energies, interatomic forces are crucial for understanding material properties and enabling simulations. To address the infinite variance problem, we evaluated existing force calculation methods and introduced the “fast warp” method. Testing across various systems demonstrates that “fast warp” is both more accurate and efficient than traditional approaches. This advancement further strengthens deep learning QMC as a promising ab initio technique.
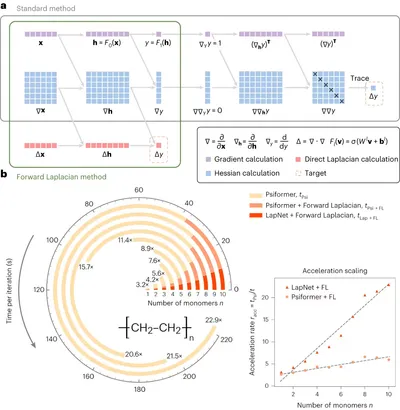
Forward Laplacian framework for deep learning QMC
The primary bottleneck in deep learning QMC training is the calculation of the wavefunction’s Laplacian, which traditionally requires computing the full Hessian matrix. Our forward Laplacian framework overcomes this by computing the Laplacian in a single forward pass, dramatically speeding up the process. We also introduce LapNet, a novel network architecture that exploits gradient sparsity and achieves O(n) acceleration without compromising accuracy. Together, these innovations expand the scalability of deep learning QMC and reduce its computational barriers, enabling broader applications.
Strongly Correlated Electronic Structure
Strongly correlated electronic systems, such as transition metal oxides, defects, and bond-breaking systems, present significant challenges for accurate theoretical modeling. These systems require highly precise quantum chemical methods to capture their complex behavior. State-of-the-art techniques, like Full Configuration Interaction Quantum Monte Carlo (FCIQMC), provide benchmark-quality data and critical insights into such strongly correlated systems, which are essential for advancing our understanding of electronic correlations in challenging materials and chemical processes.
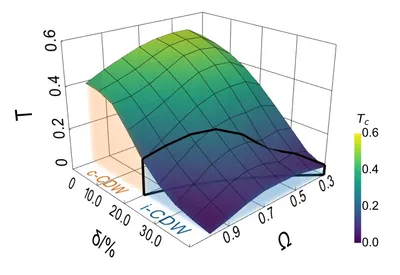
Commensurate to incommensurate transition of three-dimensional charge density waves
Although Holstein model is the simplest model containing electron-phonon interaction, its analytical solution is intractable and expensive cost prevents almost all the previous numerical studies from touching the 3D Holstein model. We develope a new program employing the fluctuation exchange approximation, based on which we determine the phase diagram of 3D charge density wave (CDW) on cubic Holstein model in the phase space spanned by phonon frequency, hole doping and temperature. The transition from commensurate CDW to incommensurate CDW is revealed, which implies that the origin of the insulating phase adjacent to superconducting phase in heavily doped bismuthates is electron-phonon interaction.
Towards an accurate electronic structure of single photon emitters in hexagonal boron nitride
We systematically examine single photon emission from defects in h-BN, critical for room-temperature quantum devices. Using FCIQMC, we determine electronic states, revealing high-spin ground states and correcting prior misunderstandings. We identify two sources for visible luminescence—including a doublet-doublet transition—shedding light on correlated electronic states and establishing a theoretical foundation for advancing quantum technologies.
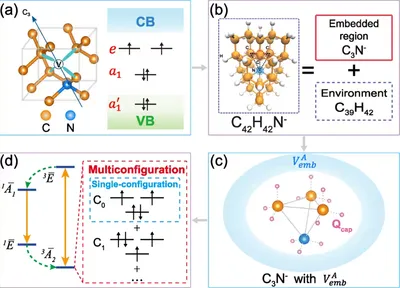
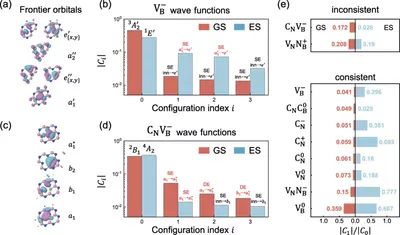
Multiconfigurational nature of electron correlation within NV-centers in diamond
To cope with the complex electron structure within NV-center, we develop a quantum embedding scheme, dubbed Density-Matrix Functional Embedding Theory (DMFET). This methods features highly accurate depiction near defect region, inherited from FCIQMC, while keeps low cost for environment regions by combining DFT. Our method sets a new benchmark for NV center states energies, and also provides a many-body view for understanding physical processes such as excitation. This work showcases a new way for calculating and understanding electronic in point defect systems.
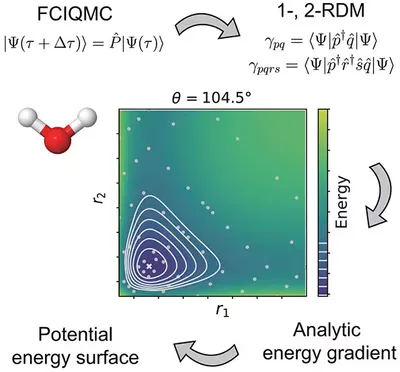
General Analytical Nuclear Forces and Molecular Potential Energy Surface from FCIQMC
We extend FCIQMC to calculate nuclear forces and potential energy surfaces, enabling unbiased sampling of force and flexible orbital selection. By combining FCIQMC with Gaussian process regression, we successfully modeled water’s potential energy surface and performed path integral molecular dynamics. Numerical tests demonstrate robust stability. This opens new avenues to explore the interplay between nuclear dynamics and electronic correlation.
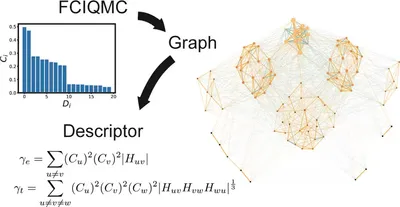
Graphic Characterization and Clustering Configuration Descriptors of Determinant Space for Molecules
FCIQMC is a powerful method for strongly correlated electronic structures, but wavefunction analysis remains limited. We introduce a graphical analysis approach, revealing that configuration clustering captures the system’s strong correlation. Two descriptors are proposed to quantify correlation strength and configuration importance, offering a new standard for selecting key configurations. This method shows strong potential for future applications in analyzing correlated systems.
Water and Carbon: Structures and Dynamics
Water and carbon are fundamental elements with complex structures and dynamics. As Prof. Enge Wang noted, “Water: soft in nature, hard in science,” its behavior in pure form, solutions, and interfaces is deeply intriguing. Carbon, with its diverse allotropes and transformations, is equally versatile. Together, they drive interdisciplinary research in physics, chemistry, materials science, energy, and environmental science, offering insights into catalysis, sustainability, and beyond.
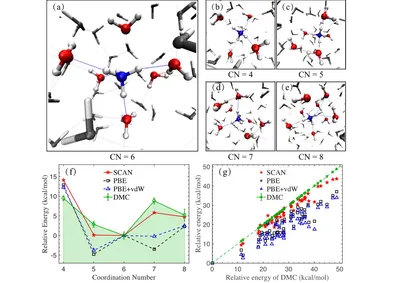
Hydration of NH4+ in Water: Bifurcated Hydrogen Bonding Structures and Fast Rotational Dynamics
Hydration dynamics of ions like NH4+ in water govern processes from crystallization to chemical reactions. Conflicting simulations and experiments left its hydration structure and rapid rotation unresolved. We merge quantum Monte Carlo and AIMD simulations to uncover bifurcated hydrogen bonds driving NH4+‘s fast rotation, reconciling theory with experiments. This QMC–AIMD framework advances modeling of aqueous systems.

Visualizing Eigen/Zundel cations and their interconversion in monolayer water on metal surfaces
Using path integral molecular dynamics, we simulate how hydrated protons (Eigen and Zundel cations) behave on Au(111) and Pt(111) surfaces. Nuclear quantum effects stabilize ordered Zundel structures, while Eigen cations form local monolayers. Simulations reveal a coupled Eigen-Zundel interconversion via interfacial proton transfer, with Pt(111) favoring Zundel configurations—unlike Au(111). These quantum-level insights advance understanding of proton dynamics in hydrogen evolution and surface-specific electrochemical processes.
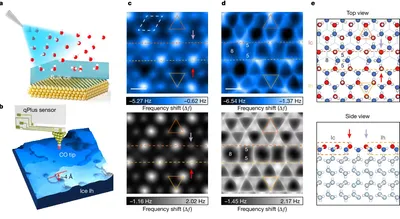
Imaging surface structure and premelting of ice Ih with atomic resolution
Ice surface dynamics, critical to premelting and atmospheric chemistry, have long been debated. DFT revealed hexagonal ice surfaces stabilize as mixed Ih/Ic nanodomains with superstructures by minimizing electrostatic repulsion. Simulations further traced premelting to defective domain boundaries, resolving structural ambiguities and advancing predictive models of hydrogen-bond-driven phase transitions.
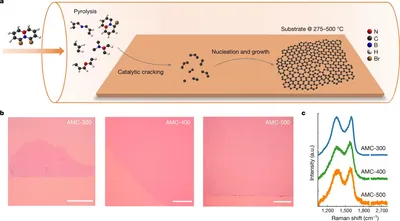
Disorder-tuned conductivity in amorphous monolayer carbon
We present a theoretical model linking atomic-scale structures to macroscopic electrical properties of amorphous monolayer carbon (AMC). We propose two key order parameters to describe the degree of disorder (DOD): the medium-range order (MRO) and the density of nanocrystallites within the AMC structure. Numerical calculations demonstrate that variations in these parameters significantly influence the material’s conductivity. Specifically, AMC films with well-defined MRO and higher nanocrystallite densities exhibit enhanced electrical conductivity, while those lacking MRO and with lower nanocrystallite densities become insulating. This theoretical framework provides a direct correlation between microstructural characteristics and the macroscopic electrical behavior of AMC, offering valuable insights for designing materials with tailored electronic properties.
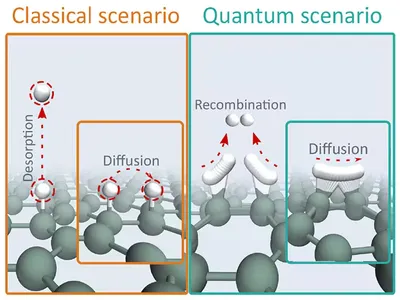
Quantum Tunnelling Driven H2 Formation on Graphene
Contrary to conventional assumptions, quantum-mechanical simulations reveal adsorbed hydrogen atoms on carbon surfaces can form H2 at low temperatures via deep tunneling. Using ring-polymer instanton theory and ab initio methods, we uncovered a multidimensional tunneling pathway—invisible to 1D models—that accelerates recombination rates by tens of orders of magnitude, reshaping predictions for H2 formation in astrochemistry and catalysis.
AI-Assisted Materials Modeling
AI-assisted materials modeling is transforming the field by combining machine learning potentials and AI-driven property prediction to accelerate material discovery. Machine learning potentials enable efficient and accurate simulations of complex systems, while AI models predict key properties like stability and conductivity directly from structural inputs. These tools reduce computational costs and unlock new possibilities for designing advanced materials, from energy storage solutions to superconductors, driving faster innovation in materials science.
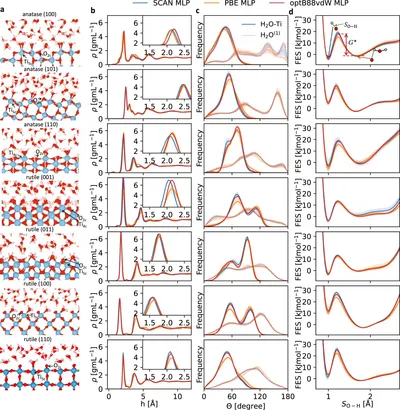
Mechanistic insight on water dissociation on pristine low-index TiO2 surfaces from machine learning molecular dynamics simulations
Water interactions with TiO2 surfaces are key for technologies like solar cells and water splitting, but most research focuses on just two surfaces. Using machine learning simulations, we study seven pristine TiO2 surfaces and find they behave differently—some break water apart, others keep it intact. These differences depend on surface structure and slab thickness, showing why simulations are crucial for understanding and designing real-world materials.
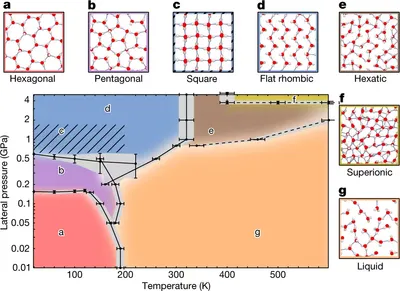
Nanoconfined water in first principles
Water in nanoscale spaces behaves very differently from bulk water, offering potential for wide applications. However, understanding its behavior is challenging due to experimental and computational limitations. Using first-principle simulations, we uncover diverse phases for nanoconfined water, including a hexatic phase and a superionic phase with high conductivity. Our findings suggest nanoconfinement could enable superionic behavior under accessible conditions, paving the way for innovative water engineering.
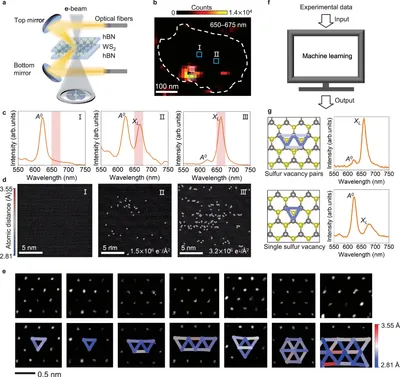
Unveiling sulfur vacancy pairs as bright and stable color centers in monolayer WS2
We employed machine learning techniques to analyze cathodoluminescence (CL) microscopy data. By applying extreme gradient boost learning algorithms (XGBoost), we effectively identified specific sulfur vacancy pairs responsible for the observed bright and stable luminescence, and linked a direct structure-property relation between defect types and CL spectra. This approach facilitated a deeper understanding of the defect-related optical properties in monolayer MoS2, highlighting the potential of machine learning in materials characterization.